Structural Basis of Ligand-gated Ion Channel Function
The biological actions of serotonin (5-HT) are mediated by at least 14 different receptors in seven different families (5-HT1 to 5-HT7). Unlike the other 5-HT receptors, which are G-protein coupled receptors (GPCR's), the 5-HT3R is a ligand-gated cation channel. The 5-HT3R has been implicated in a number of physiological and pathophysiological processes. 5-HT3R antagonists such as granisetron have selective antiemetic activity when nausea is induced by anesthesia, cytotoxic chemotherapy, or radiation. This activity allows patients to tolerate higher doses and longer treatment with chemotherapy agents, and, along with the prevention of post-operative nausea and vomiting, represents the main therapeutic use of 5-HT3R antagonists. 5-HT3R antagonists, however, are not without side effects, including arrhythmias. As these side effects are most likely mediated through interactions with other targets, there is a need for more selective antagonists, which is where elucidation of the structural features of the ligand-binding domain may provide useful information.
Ligand-gated ion channels such as the nicotinic acetylcholine receptor (AChR), the GABA type A (GABAAR) and type C (GABACR) receptors, the glycine receptor (GlyR), and the 5-HT3R play important roles in signaling in the central and peripheral nervous systems, and are targets for a number of therapeutic agents. These receptors exhibit a great deal of homology between each other and are considered to be members of a single gene family- the cys-loop ligand-gated ion channel (LGIC) family. The members of this family are transmembrane proteins that transduce the binding of a small-molecule agonist into a series of conformational changes that ultimately result in the opening of an ion-selective channel. A central question concerning cys-loop LGIC's is what is the architecture of the ligand-binding site, and how does agonist binding to a site far removed from the ion channel induce channel opening?
We use an approach that combines molecular biology, biochemistry, electrophysiology, and (eventually) structural biology to analyze the structural basis of ligand-gated ion channel function, in particular the architecture of the ligand-binding domains. A series of well-defined, mutant channels or receptors, which differ from the wild-type by one or two amino acids, are generated using site-directed mutagenesis. The mutant receptors are then expressed in transfected mammalian cells, where their properties are studied using ligand-binding and electrophysiological assays. Detailed comparison of the functional properties of the mutant receptors with those of the wild-type can elucidate the structural features that underlie various aspects of ion channel or receptor function and expression. Our current studies focus on architecture of the ligand-binding domain of the 5-HT3R. A great deal of work has been done on the nicotinic AChR by a number of different laboratories, and important structural details have emerged concerning the transduction of agonist binding to AChR gating. However, it is not clear to what extent these features are conserved among other members of the cys-loop LGIC family, and so we have focused our efforts on another member of the gene family, the 5-HT3R, in order to determine common vs receptor-specific aspects of the transduction process.
The 5-HT3R exists in both homopentameric (consisting of only the 5-HT3A subunit) and heteropentameric (containing the 5-HT3A and 5-HT3B subunits). Analysis of the expression patterns of the 5-HT3A and 5-HT3B subunits has led to the suggestion that the bulk of the 5-HT
Molecular Modeling of Ligand-Receptor Complexes
Although atomic-resolution structures are not available for members of the eukaryotic cys-loop LGIC family, the structure of soluble acetylcholine binding proteins (AChBP's) from several mollusks (Lymnaea, Aplysia, Bulinus) complexed with various ligands have been determined at atomic resolution. The AChBP has a high degree of homology to the extracellular domain of the AChR and other cys-loop LGIC's, and is considered to be an excellent model for the three-dimensional structure of the extracellular domain of all members of the cys-loop LGIC family. We have used the three-dimensional structure of the AChBP from various species complexed with different ligands as templates to create a structural model of the ligand-binding domain of the 5HT3R in the apo, agonist-bound, and antagonist-bound states using using MODELLER followed by side-chain refinement using a backbone-dependent rotamer library (SCWRL). Shown below are top and side views of the pentameric complex in the apo (unliganded) form, with each subunit shown in a different color.
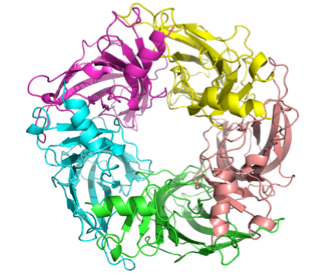
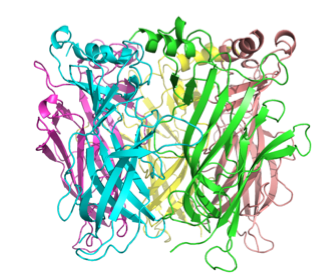
The ligand-binding sites are
at the subunit-subunit interfaces. We can then use this
model to carry out ligand-docking simulations to produce a
structure of a ligand-receptor complex using
AutoDock.
Double-Mutant Cycle
Analysis of Ligand-Receptor Interactions
Double-mutant cycle anlaysis can be used to determine which
residues in the receptor interact with specific parts of
the ligand. The underlying logic is that if residue x
interacts with region y of the ligand, the effect of
changing y on ligand-receptor interactions should be
affected by mutation of x in the receptor. A free energy of
interaction ΔΔGint, is calculated from the Kd or Ki
values obtained from analysis of the interaction of a
series of ligands with wild-type and mutant receptors.
A ΔΔGint value different than 0 is consistent
with the notion that the portion of the ligand that has
been changed makes a physical interaction with the amino
acid residue that has been mutated.
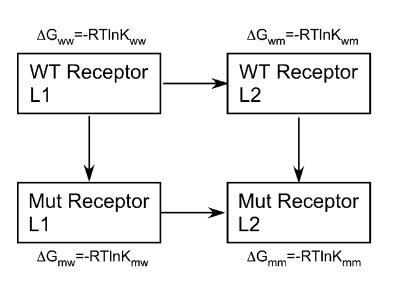
ΔΔGint=(ΔGww-ΔGwm)-(ΔGmw-ΔGmm)
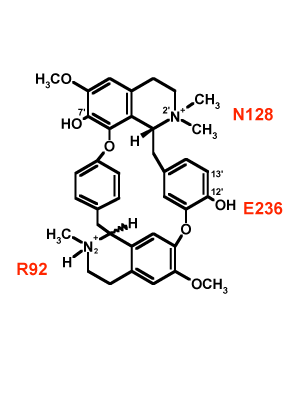
We have used the rigid competitive
antagonist d-tubocurarine (dTC) to determine specific
points of interaction between residues in the
ligand-binding domain and portions of the antagonist. Using
a panel of dTC analogs we have mapped three different
residues (R92, N128, E236) onto three different portions of
dTC.Since dTC is a rigid molecule, these data provide
information on the spatial separation between these
residues, which can then be used as parameter restraints in
MODELLER. Shown below is a docked dTC molecule in the
ligand-binding domain of the
5-HT3R
using the spatial data obtained form the double-mutant
cycle analysis.
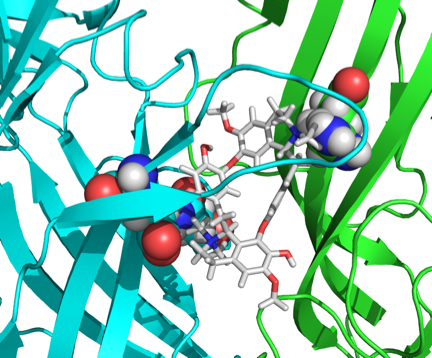
Mapping additional residues in the ligand-binding site onto
dTC or other rigid ligands offers the possibility to
determine the architecture of the ligand-binding domain in
the absence of X-ray crystallographic structures, and may
provide guidance for the design of novel therapeutic agents
targeting the 5-HT3R.